역학(疫學)이란 인구집단에서 병원 원인을 연구하는 학문이다. 물리학의 한 분야인 力學, 주역의 괘를 해석하여 음양 변화의 원리와 이치를 연구하는 학문인 易學(이것이 진정한 학문인가?)과는 다르다. 우연히 발견한 글인 역학자가 노벨상을 받을 수 없는 이유도 한번 읽어보자.
감염병의 역학조사와 병원체 genome sequence를 어떻게 연결하여 감염성 질환에 대처할 것인가? 이에 대해서는 2014년 Genome Biology에 실렸던 리뷰 논문인 Epidemiologic data and pathogen genome sequences: a powerful synergy for public health를 참조하자. 이 논문에 의하면, 감염병을 연구하는 역학자의 기본 질문은 이러하다.
- Is there an outbreak?
- Where, when and how did a pathogen enter the population of interest?
- How quickly is the number of infections from the pathogen growing (that is, what are the epidemic dynamics)?
- How is the pathogen spreading through the population?
- What genes or genotypes are associated with the pathogen's virulence or other phenotypes of interest?
Pathogen genomics는 이러한 질문에 대해 답을 할 수 있는 매우 중요한 근거를 제시한다. 물론 오늘의 포스팅에서는 더 깊게 논의하지는 않겠다.
내가 다수의 병원체 샘플에 대한 whole-genome sequencing data를 이용한 분석, 즉 genotyping(SNP-based라고 좁혀서 이야기해도 좋다)과 resistance gene finding 등을 실무에서 처음 접하게 된 것은 작년 하반기였다(참고로 Sanger sequencing은 1991년 무렵, 대용량 genome sequencing은 2000년부터 해 왔음). 당시에는 결핵균의 일루미나 시퀀싱 raw data가 출발점이었다. Reference mapping 기반의 방법과 assembly 기반의 방법 사이에서 고민을 하다가 결국은 일본에서 개발한 TGS-TB(Total Genotyping Solution for Mycobacterium tuberculosis, 현재 버전 2)라는 매우 유용한 웹사이트를 찾아내는 것으로 일단락되었다. 그러나 이 작업 중에 CLC Genomic Workbench의 Microbial Genomics Module(typing and epidemiology 기능)을 구입하게 되었고, Sanger Institute의 Roary라는 도구도 알게 되었다.
Roary는 원래 pan genome 분석을 위한 도구이다. 유전자의 염기서열을 포함하는 GFF3 파일을 input data로 하여(즉 annotated genome sequence가 필요) core gene과 accessory gene을 집계하고, 이로부터 multiple sequence alignment까지를 해 준다. 다양한 2차적인 플롯을 만들 수 있는 중간 결과물도 제공하는 것이 특징이다. 단, sequencing raw data는 다루지 못한다. SNP을 추출하거나(snp-sites) phylogenetic tree를 그리는 일(fasttree)은 별도로 실시해야 한다.
다음으로 알게 된 소프트웨어는 매릴랜드 대학교의 Harvest suite이다. 이것은 core genome alignment와 visualization까지를 할 수 있다. 입력물은 assembled genome sequence로서 annotation은 필요하지 않다. 분석에 투입할 유전체는 >=97% ANI를 충족하는 가까운 것들이어야 한다. MUM에 기반한 locally collinear block(LCB) 계산 및 SNP 검출을 해 주기에 실행 속도가 매우 빠른데, Parsnp(rapid core genome multi-alignment) 커맨드 한 줄로 모든 분석이 끝난다. 더욱 좋은 것은 Gingr이라는 visualization tool이 포함된다는 점이다. 다음은 시험적으로 생성한 분석 결과이다. 왼쪽에는 SNP-based tree가, 오른쪽에는 SNP map이 보인다.

Gingr은 standard alignment format을 수용하는 일반적인 용도의 viewer로도 쓰일 수 있다. 패키지에 포함된 harvesttools를 병용하면 사용하여 다음과 같은 다양한 파일 포맷간의 전환이 가능하다.
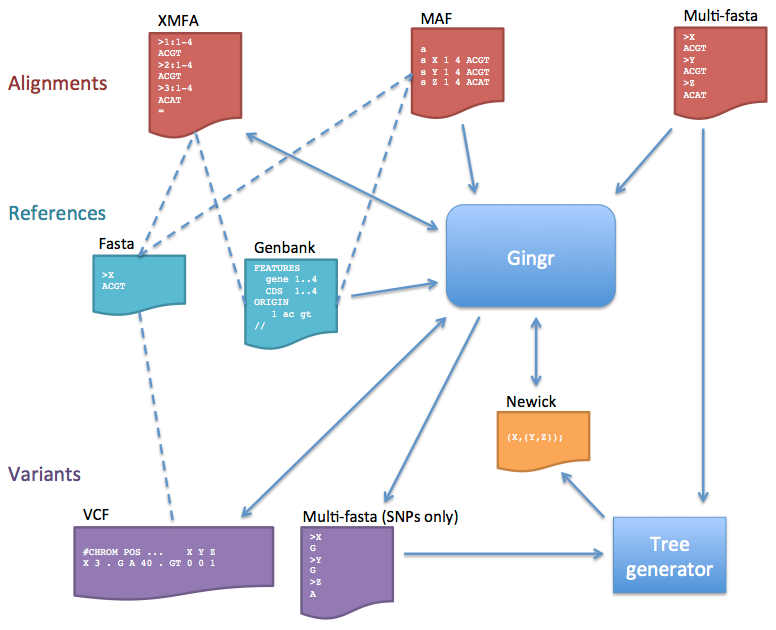 |
| 그림 출처: http://harvest.readthedocs.io/en/latest/content/gingr/types.html |
마지막으로 살펴볼 것은 North Arizona SNP pipeline, 즉 NASP이다. 이 도구는 raw sequencing read와 assembly를 가리지 않고, 다양한 aligner와 SNP caller를 활용할 수 있으며, monomorphic 및 polymorphic site를 검출 가능하고, job management system을 채용하는 것을 특징으로 한다. NASP는 2016년에 bioRxiv에 먼저 공개되었다가 신생 학술지인 Microbial Genomics(MGen)에 같은 해 출간되었다. 그러나 내 눈에 뜨인 것은 이틀전이다.
만약 long read data가 input data라면 어떻게 할 것인가? 가능하다면 오류가 적은 genome assembly를 만들어서 적당한 pipeline에 투입하는 것이 바람직할 것이다. 이제는 long read를 만들어내는 기술이 PacBio이 유일한 것이 아니라 Oxford nanopore sequencing까지 늘어났으니 점점 공부할 것이 많아진다...
댓글 없음:
댓글 쓰기