어쨌든 usearch는 LS-BSR의 기본 클러스터링 도구이다. 다른 대안으로는 vsearch, cd-hit 등이 있다. 서열 클러스터링은 왜 하는가? 유사한 서열을 묶어서 데이터 분량을 줄이는 것이 주된 목적 중의 하나이다. Non-redundant sequence set을 작성하거나, 방대한 metagenome read를 처리하여 그 분량을 줄이는 것이 주된 application이다. 묶인 서열 클러스터에 대하여 하나씩의 대표 서열(representative sequence; usearch에서는 "centroid"라는 표현을 쓰는 것 같다)을 선정하거나, 혹은 consensus sequence를 만들어낼 수 있다. 아니면 클러스터에 속하는 모든 멤버 서열들을 그냥 나열하는 것도 가능하다.
그동안 상습적으로 blastclust를 써 왔는데 왠지 시대에 뒤떨어진 것 같다는 느낌이 들어서 오늘은 usearch를 살짝 사용해 보았다. 나와 비슷한 생각을 가진 사람이 또 있었는지 Biostars에 다음과 같은 글이 있다. 현재의 blast+ 패키지에는 blastclust가 빠져 있으니 그럴 생각이 들 만도 하다.
Questions: Blastclust has been deprieciated. Does anyone know why? (depreciated를 deprecated로 읽었다)Usearch는 high-througput search & clustering 도구로서 Robert C. Edagr 박사가 개발하였다(웹사이트; usearch home 링크). 원래 그는 입자물리학 전공자로서 기업을 거쳐 2001년부터는 독립 과학자('independent scientist')로 일하고 있다. Usearch는 32 비트 버전의 경우 비상업적 용도로는 무료(소스 코드는 비공개), 64 비트 버전은 유료이다. 다중서열정렬 프로그램으로 유명한 muscle도 그가 개발하였다.
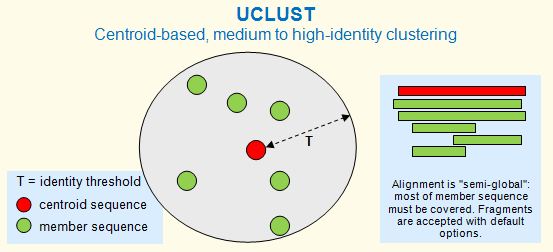
Usearch는 몇 가지의 클러스터링 알고리즘을 내장하고 있는데, 그중에서 usearch -cluster_fast 혹은 usearch -clust_smallmem 명령으로 기동되는 uclust에 대해서 좀 더 알아보자. 다음 그림은 uclust의 알고리즘을 한 눈에 잘 들어오게 보여준다(UCLUST algorithm). 웹사이트의 설명을 인용하자면 "A cluster is defined by one sequence, known as the centroid or representative sequence"이다. Ucluster는 단백질의 경우 identity ~50% 이상, 핵산의 경우 ~75% 이상인 서열에 적용하는 것이 바람직하다. Usearch가 제공하는 클러스터링 관련 모든 방법은 여기를 참고하라. Uclust는 2010년 Nucleic Acids Research에 "Searching and clustering orders of magnitude faster than BLAST"라는 논문으로 발표된바 있다(링크). 논문의 저자는 Robert C. Edgar 1인이다. 곳곳에서 'I'를 주어로 시작하는 문장이 보인다. 멋지지 않은가? 2002년 Genome Research에 실렸던 W. James Kent의 "BLAT-The BLAST-Like Alignment Tool" 논문(링크)도 그렇다.
 |
| https://www.drive5.com/usearch/manual/uclust_algo.html |
클러스터 set는 다음과 같은 기준으로 만들어진다.
- 모든 centeroid의 서로에 대한 similarity는 T보다 작다.
- 클러스터 내 멤버 서열의 similarity는 centroid에 대하여 T와 같거나 크다.
OTU(operational taxonomic unit) clustering은 특별한 문제를 야기한다고 한다. Length sort를 하게되면 biological outlier를 centroid로 만드는 부작용이 유발되고, 종과 속을 다른 클러스터로 쪼개어서 alpha diversity를 뻥튀기하는 현상이 생긴다. 상세한 내용은 매뉴얼 페이지 혹은 2010년도 Bioinformatics에 실린 논문 "Search and clustering orders of magnitude faster than BLAST"를 참조하라.
Usearchf를 이용한 클러스터링의 기본 실행법은 다음과 같다(USEARCH command line).
usearch -cluster_fast seqs.fasta -id 0.9 -centroids nr.fasta -uc clusters.uc화면으로 출력되는 메시지를 살펴보자.
00:00 42Mb 100.0% Reading HB03_20180914.ffn 00:00 8.7Mb Pass 1...2408 seqs, 2393 uniques, 2382 singletons (99.5%) 00:00 367Mb Min size 1, median 1, max 3, avg 1.01 00:00 370Mb done. 00:00 370Mb Sort length... done. 00:18 399Mb 100.0% 2391 clusters, max size 3, avg 1.0 00:18 399Mb 100.0% Writing centroids to HB03_nr.fasta 00:18 399Mb 100.0% Writing clusters Seqs 2393 Clusters 2391 Max size 3 Avg size 1.0 Min size 1 Singletons 2378, 99.4% of seqs, 99.5% of clusters Max mem 400Mb Time 18.0s Throughput 132.9 seqs/sec.
두번째 줄에 표시된 수치는 이해하기가 좀 어렵다. 메시지 후반에 나온 확정된 수치를 참고하거나, 혹은 클러스터를 FASTA file로 출력하여 직접 서열의 수를 세는 것이 더 확실할 것이다. 2393개의 (unique) sequence라는 것도 어떻게 계산된 것인지 잘 모르겠다. Singleton이라 하면 클러스터링 후 멤버가 하나인 서열을 의미하는 것일텐데, 이것과 unique는 무엇이 다른 것일까?
-cluster_fast는 command, -centroids는 output option이다. 입력 FASTA file은 -cluster_fast 바로 뒤에 와야 한다. 클러스터 정보는 clusters.uc 파일에 기록된다. 이 파일의 포맷은 ucluster의 매뉴얼을 참조하라(링크). 클러스터를 구성하는 서열을 얻고 싶다면 다음과 같이 -cluster 옵션을 주어야 한다(cluster_fast에서만 작동). 그러면 cluster_dir 아래에 c_0, c_1, c_2...과 같은 서열 파일이 생긴다. 각 파일은 하나의 클러스터를 의미하며, 파일 내의 첫번째 서열이 바로 centroid가 된다. cluster_dir 디렉토리는 usearch 실행 전에 만들어 두어야 한다.
-cluster_fast는 command, -centroids는 output option이다. 입력 FASTA file은 -cluster_fast 바로 뒤에 와야 한다. 클러스터 정보는 clusters.uc 파일에 기록된다. 이 파일의 포맷은 ucluster의 매뉴얼을 참조하라(링크). 클러스터를 구성하는 서열을 얻고 싶다면 다음과 같이 -cluster 옵션을 주어야 한다(cluster_fast에서만 작동). 그러면 cluster_dir 아래에 c_0, c_1, c_2...과 같은 서열 파일이 생긴다. 각 파일은 하나의 클러스터를 의미하며, 파일 내의 첫번째 서열이 바로 centroid가 된다. cluster_dir 디렉토리는 usearch 실행 전에 만들어 두어야 한다.
usearch -cluster_fast seqs.fasta -id 0.9 -clusters cluster_dir/c_Usearch는 기본적으로 입력 파일에 존재하는 서열의 순서에 따라서 일을 한다. Fragment를 포함시키는 속성에 의하여 이것이 centroid로 쓰이게 되면 결과적으로는 매우 바람직하지 못하다. 따라서 -sort length 옵션을 적용하는 것이 좋다(UCLUST sort order). 다른 sort 옵션으로는 abundance가 있다. 정렬 순서는 큰 것에서 작은 순이다.
그러면 2016년에 발표된 vsearch(논문 링크; GitHub)는 무엇인가? 소스코드가 공개되어있지 않고 알고리즘 설명이 빈약한 usearch를 대신하고자 만든 open source tool이라고 한다.
마지막으로 CD-HIT를 빼놓을 수 없다(GitHub). 거대한 분량의 데이터를 초고속으로 다루는 cd-hit에는 다양한 핵심 프로그램과 스크립트가 포함되어 있다.
추가 작성: 2018년 10월 23일
위의 내용은 2018년 10월 23일에 대폭 수정되었다. 더불어서 새로 조사한 사항도 추가하고자 한다. 2011년도에 발표된 SiLiX 패키지라는 클러스터링 도구가 있었는데(링크) 크게 인기를 얻지는 못한 것 같다. 이를 개발한 프랑스에서는 꾸준히 사용되는 것 같다. 논문에서는 usearch나 uclust를 언급하지는 않았다.
프로그램 실행 방법을 소개한 리뷰 글 "How to cluster protein sequences: tools, tips and commands"도 소개해 둔다(링크).
댓글 없음:
댓글 쓰기