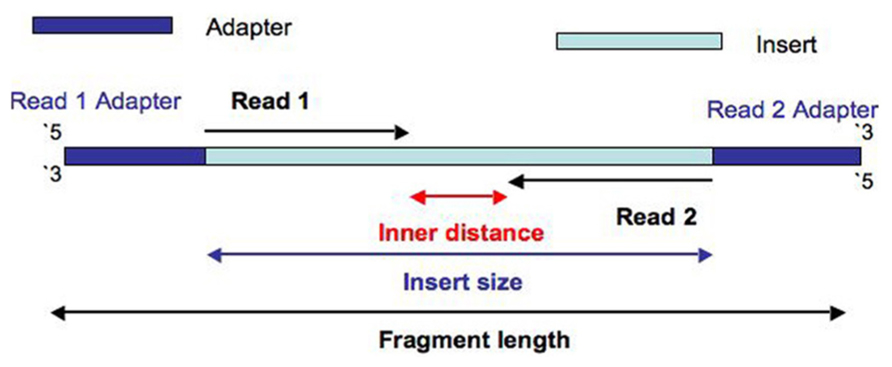
ART sequencing simulator의 여러 파라미터 중 라이브러리의 크기와 직접 관련이 있는 것은 '-m'(the mean size of DNA/RNA fragments for paired-end simulations)이다. 길이 분포는 표정규분포를 가정하되 표준편차는 '-s' 파라미터로 설정한다. 일루미나 시퀀싱 라이브러리의 insert size와 fragment size는 보통 다음 그림과 같이 서로 다르게 정의된다.
 |
| 출처: BioStars |
정리하면 다음과 같다(참조: BioStars).
- insert size = the sequence between adapters
- fragment size = insert size + adapters
Fragment length는 양 끝의 어댑터를 포함하는 길이에 해당한다. CLC Genomics Workbench의 mapping 또는 de novo assembly report에서 만들어지는 paired reads distance 분포는 위 그림에서 inner distance에 해당할 것으로 생각하기 쉬운데, CLC Genomics Workbench 매뉴얼에 의하면 'The paired read distance includes the full read sequences"라고 명시되어 있다. 따라서 위에서 보인 그림의 insert size에 해당한다. 만약 이렇게 하지 않으면 forward와 reverse read가 서로 겹치는 경우 inner distance는 음수의 값을 갖게 될 것이다.
art_illumina의 '-m' 파라미터는 fragment size라고 하였으니 양 끝 어댑터의 길이를 합친 값을 제공해야 할 것처럼 보인다. 그런데 read simulator의 입장에서 어댑터의 크기를 고려할 일이 있을까? 몇 번의 시행착오를 거친 끝에 '-m' 뒤에는 insert size(CLC Genomics Workbench의 paired read distance 대푯값)에 해당하는 숫자를 넣으면 된다는 것을 알았다. 오랫동안 art_illumina를 사용해 왔지만 simulation으로 만들어진 read의 실제 분포를 측정해 본 일은 없었다. 대충 '-m 400' 정도로 놓고 read를 만들어서 사용하기만 했었다.
다음은 실제 데이터인 SRR1144835의 자료를 샘플링하여 CLC Genomics Workbench에서 de novo assembly를 한 결과 리포트에서 딴 것이다. 분포가 예쁘지는 않지만 대략 470 bp 정도를 목표로 하였다.


Torsten Seemann의 블로그 "The Genome Factory"에 paired-end read의 길이에 대한 명쾌한 설명이 나온다. 이미 내 블로그에서 이 링크를 과거에 한 번 소개했었던 것 같다. 오늘 발견한 문제는 ART나 CLC에서 다루는 길이 관련 정의가 일반적으로 받아들여지는 것과는 조금 다르다는 것이다.
댓글 없음:
댓글 쓰기