Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol 2000; 68:461-466 PubMed PDF 파일
흔히 IJSEM이라고도 줄여서 부르는 학술잡지 "International Journal of Systematic and Evolutionary Microbiology"는 미생물의 분류학을 다루는 전문지이다. 새로운 분류군을 보고하고 분류학의 기준을 제시하는 역할을 한다. 단순히 미생물이라고 하면 무척 다양한 생물들이 포함된다. 예를 들어서 광합성을 하는 단일세포 생물인 클로렐라도 넓은 의미의 미생물에 속한다. 그러나 IJSEM은 세균(bacteria)과 효모(yeast)만을 다룬다. 이 글에서 말하는 좁은 의미의 미생물은 핵이 없는 원핵생물(prokaryote), 즉 박테리아(세균)를 뜻한다. 엄밀히 말하면 고세균(archaea)도 원핵생물의 한 부류이지만 말이다.
신종으로 공인이 되려면 이에 대한 표준 균주(type strain)를 공인 균주 은행 최소 두 곳에 이를 기탁하여 균주 번호를 받아야 한다. 우리나라에는 한국생명공학연구원에서 운영하는 전북 정읍 소재 생물자원센터(KCTC, Korean Collection for Type Cultures 링크)가 있다. 균주 번호 끝에 윗첨자 T(혹은 괄호를 둘러친 T)가 있다면 이것은 그 종의 표준 균주임을 뜻한다.
박테리아는 형태적으로 비교적 단순하여 그 모습을 보고 분류를 하기가 대단히 어렵다. 따라서 형태적 특성 외에도 분석적 방법(지질 분석, 전체 세포 단백질 분석)과 유전적 방법(G+C 비율, DNA sequence...)을 총동원하게 된다.
두 생명체가 같은 종에 속하는지를 판별하는 문제 역시 박테리아에서는 쉽지 않다. 다른 생물에서는 서로 교배를 해서 생식 가능한 자손을 낳을 수 있으면 동일 종으로 정의한다. 그러나 박테리아는 유전물질을 서로 1:1로 교환하는 이러한 과정 자체가 없다(파트너에게 유전물질의 일부를 일방적으로 주입하는 과정은 있다). 그러나 두 개체 혹은 두 집단이 동일 종에 속하는지를 판별하기가 어려운 것이다.
전통적으로 가장 많이 쓰이는 기준은 DNA-DNA hybridization(DDH)을 하여 70%를 넘는 similarity가 나오면 동일 종으로 판별한다는 것이다. IJSEM에 새로운 미생물 종을 보고하려면 여러 특성을 조사하는 것 외에 알려진 다른 종의 표준 균주에 대하여 DDH가 70% 이하로 나와야 한다. 문제는 이 실험 기법이 숙련되기가 어렵고 재현성도 높지 않다는 것이다. 나 역시 이 실험을 해 본 적도 없고, 이 실험을 하는 모습을 옆에서 구경한 적도 없다. 신종 미생물 등록 실적에서 세계 1등이라 해도 과언이 아닌 성균관대 윤정훈 교수가 근처에서 근무할 때 구경이라도 한번 할 것을 그랬다.
상황이 이러하니 과거에 부정확한 DDH 실험 결과를 가지고서 신종으로 보고했던 미생물이 나중에는 이미 알려진 종과 같은 것이라는 논문(A is a later heterotypic synonym of B...)이 종종 발표되는 것이다. 이제는 NGS를 통해서 genome sequencing을 누구나 싼 값에 할 수 있는 시대가 되었다. 따라서 까다로운 DDH 실험을 할 필요성이 점점 줄어드는 것이다. DNA 간의 유사도는 결국 염기서열 배열 정보에 기인하는 것이므로, 유전체 DNA를 읽어서(이 과정은 철저히 표준화가 되었고 오차나 재현성의 문제가 거의 없다) 서로 비교하면 DDH를 대체할 수 있는 것이다. 물론 16S rRNA gene sequence도 균주 동정에서 매우 중요한 지표로 쓰이고 있지만 이것이 98.7% 보다 같거나 높으면 두 균주가 동일한 종인지 혹은 아닌지를 알기 어려워서 DDH 실험을 해야만 했다.
서론이 길었다. 오늘 소개하는 논문에서는 이러한 유전체 데이터를 원핵생물의 분류학에서 쓰고자 할 때 준수해야 할 최소 기준을 제안하는 것이다. 아직 IJSEM의 논문 투고 요령에는 신종 보고 논문을 투고할 때 반드시 유전체 시퀀싱을 완료하여 공개하라는 요구사항은 없지만 시퀀싱에 드는 비용이 점점 낮아지는 추세이므로 이것이 포함되는 것은 시간 문제일 것이다.
이 논문이 발표된 이후 미생물 유전체 해독 서비스를 주업무로 하는 천랩에는 이에 대한 문의와 불평이 쏟아지고 있다는 말을 들었다. 연구자들이 과거에는 하지 않던 일을 새로 해야 하니 부담이 될 수는 있을 것이다. 하지만 세계적인 추세를 거스를 수는 없고, 남들이 믿고 의지할 수 있는 정확한 미생물 신종 보고를 하려면 반드시 필요한 일이라고 생각한다. 실제 유전체 해독을 하면서 해당 종의 표준 균주 유전체 정보가 부족하여 애를 먹는 일이 많기 때문이다. 많은 연구자가 이 논문의 취지를 받아들인다면 천랩으로서는 더욱 사업이 확장되는 계기가 될 것이다. 논문의 저자 중 천랩의 중요 관계자가 있다는 것은 애교로 봐 주자.
두 미생물 유전체의 similarity 혹은 distance를 산출하는 프로그램은 꽤 많은 종류가 있다. 이를 통틀어서 OGRI(overall genome related index)라고 하며, 오늘 소개하는 논문의 표 1에 OGRI 계산 도구를 소개하였다. 가장 널리 쓰이는 수치인 ANI(average nucleotide identity)는 95~96%일 때 DDH 70%에 해당한다. ANI의 이론은 단순하지만 실제 구현하는 방법은 다양하다. 유전체를 일정 길이로 잘라서 쓸 것인가 혹은 유전자 영역만 추출하여 쓸 것인가? Sequence alignment에는 blast를 쓸 것인가 또는 mummer를 쓸 것인가?
OGRI는 종 수준의 판별에는 매우 유용하지만 subspecies와 genus(혹은 그 상위 레벨)의 판별에 대해서는 아직 객관적인 기준을 제시하기 어렵다. 하지만 논문에서는 이에 대한 아이디어를 제시하고 있다.
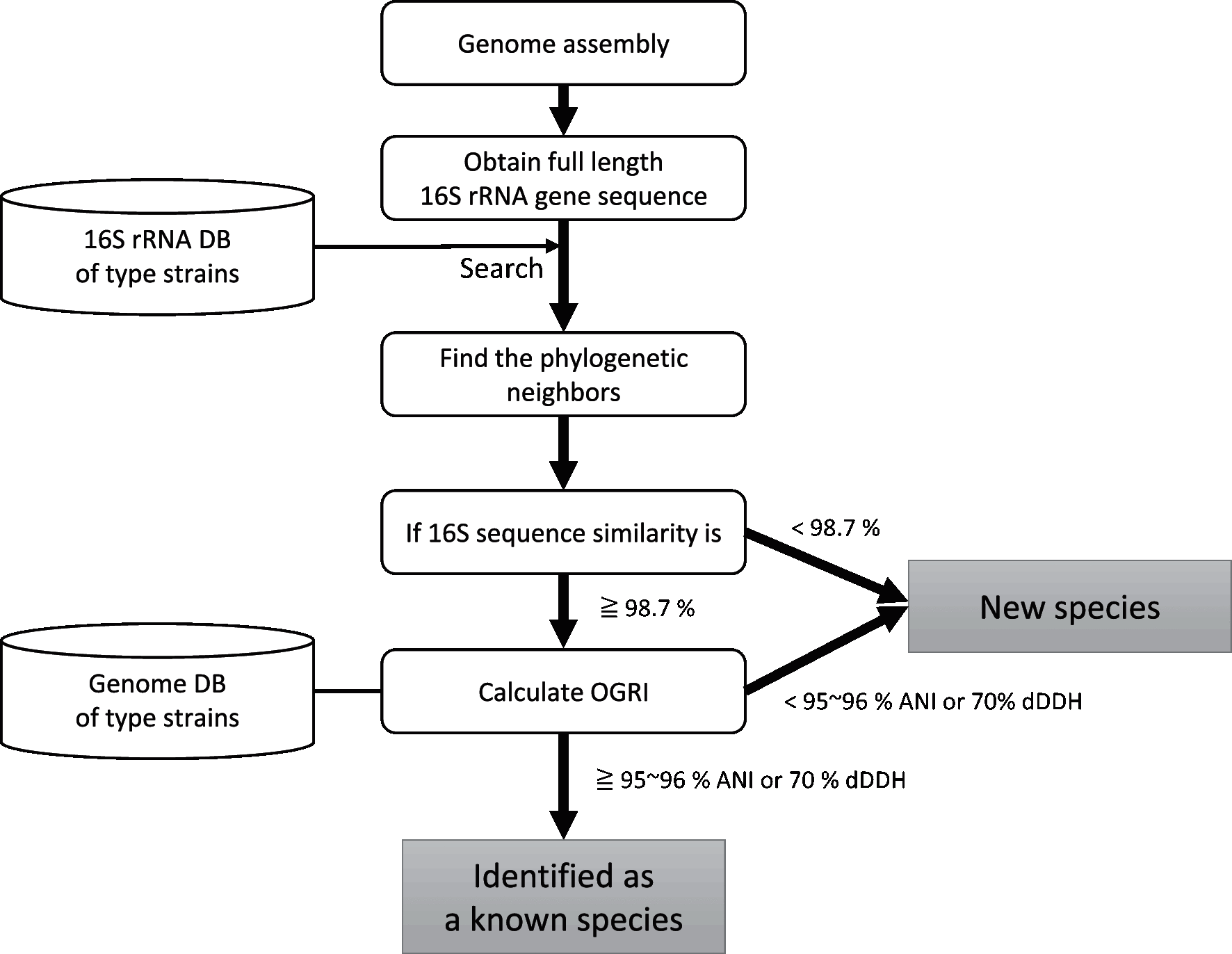 |
| 그림 1. Workflow of genome based classification at the species level. To recognize new genera, phylogenomic treeing should be used. 출처: http://ijs.microbiologyresearch.org/content/journal/ijsem/10.1099/ijsem.0.002516#tab2 |
그러면 논문의 결론에 해당하는 "minimal standard"가 무엇인지 나열해 보자.
- DNA sequencing platform: 무슨 장비를 사용하였는가?
- Quality of raw NGS data and assembled genome sequences: Genome size, the number of contigs and N50, sequencing depth of coverage
- Authenticity of the genome assembly: genome assembly에서 발견된 16S rRNA gene sequence를 실제 보고된 유사종의 그것과 맞추어 보는 것이 중요하다. 알려진 종과 너무 유사하여 16S rRNA 서열이 너무나 비슷하다면, gyrB, rpoB, recA와 같은 단백질 코딩 유전자 서열을 사용하라.
- Contamination in the genome assembly
4번의 문제는 의외로 심각하다. 범주는 약간 다르지만 샘플 표지를 잘못하였거나 균주 자체를 잘못 동정한 상태로 만들어진 유전체 어셈블리를 GenBank에 올리는 일도 있다. 그리고 일루미나의 시퀀싱 키트에 포함된 phiX174 콘트롤 DNA가 시퀀싱되어서 최종 결과물에 남는 일도 요즘 종종 발견된다. 원래 정상적인 겨우 이 서열 정보는 최종 결과에는 남아서는 안된다.
미생물 분류학은 자유로움보다는 엄격한 기준 준수를 요구하는 독특한 학문 분야이다. 공인되지 않은 종 이름은 사용하기도 어렵다. 하지만 생물계에서는 '오늘부터 나는 너하고 다른 species이니 그리 알아라'하는 일은 절대로 벌어지지는 않는다. 이러한 자연세계를 대상으로 인간이 가장 잘 하는 일이 형태를 구분하여 무리를 짓고 편을 나누는 것이다. NGS라는 새로운 기술이 보급되어 미생물 분류학도 이렇게 변화해 나가고 있다.
댓글 없음:
댓글 쓰기