요즘 시대에 새롭게 분리한 미생물을 동정하는 가장 빠르고도 확실한 방법은 아마도 유전체 해독일 것이다. 이미 공개된 type strain(표준균주)의 유전체와 비교하여 ANI가 일정 threshold 보다 크면(대략 95% 이상이며, 종마다 조금씩 다르게 정의한다고 알려짐) 그 종에 속한다고 판명하면 된다. 그동안의 문제는 이 과정을 빠르게 진행하기 어려웠다는 것이다. 대충 어느 genus에 속하는지 알게 되면 여기에 속하는 미생물의 공개된 유전체(특히 공인된 종의 표준균주 포함)를 전부 받아서 pyani나 fastANI로 분석을 해야 하는데, 많은 시간이 소요된다. 16S rRNA 분석에 의해 대충 어느 종에 가까울 것이라고 판단하여 후보 종을 줄여 나갈 수도 있으나, 재수가 없으면 비교 대상을 더 넓혀야 한다. 특히 Bacillus나 Paenibacillus와 같은 대책이 없는 genus의 경우 XYZbacillus라고 이름이 붙은 녀석들까지 건드려 봐야 할지도 모른다.
현재 22,176개의 type strain 정보를 갖고 있는 DSMZ의 Type (Strain) Genome Server, 즉 TYGS도 좋은 자원이지만 웹사이트에 유전체 서열 파일을 제출하고 한참을 기다려야 한다. 50개 정도 유전체 파일을 밀어 넣으려면 시간도 오래 걸리고 상당히 미안해진다. RAST server에 작업을 제출할 때에는(꽤 오랜 전의 일이 되었지만) 별로 미안한 느낌이 없었는데 말이다.
1990년대에 Jean P. Euzéby가 시작하여 LPSN이라는 약자로 잘 알려진 List of Prokaryotic names with Standing in Nomenclature가 DSMZ로 넘어가면서 TYGS는 미생물 명명법 체계와 유전체 정보에 기반한 더욱 좋은 자원이 되었을 것이다.
호주 퀸즐랜드 대학에서 개발한 GTDB(Genome Taxonomy Database)는 마커 유전자(박테리아: 120개, 고세균: 53개)를 이용하여 계통수를 구성하고, ANI를 이용하여 종 구분을 하여 구축한 유전체 기반 미생물 분류 DB라고 할 수 있다. 다시 말해서 웹 인터페이스에서는 이미 구축해 놓은 정보(2024년 4월 24일자 공개버전에서는 596,859개 유전체 수록)를 보여주는 것과 FastANI calculator를 제공한다.

만약 수십 건 정도의 유전체를 이용하여 정확한 종 분류를 하고 싶다면 GTDB-Tk를 로컬 컴퓨터에 설치하면 된다. 이에 대한 논문 2020년에 별도로 나왔다(링크). Query genome에 대한 taxonomic classification 세부 사항은 논문을 직접 확인하는 것이 좋을 것이다.
요즘은 업무를 하는 틈틈이 10여년 전에 일루미나로 시퀀싱해 둔 72개의 미생물 유전체 정보를 선별하여 국가 바이오 데이터 스테이션에 등록하기 위한 작업을 진행 중인데, 시퀀싱 당시에 붙여놓은 각 균주의 종 분류가 올바른지 확인하기 위해서 GTDB-Tk를 이용하기로 하였다. 이에 관한 상세한 스토리는 72 prokaryotic genomes에 작성해 나가고 있다.
Bioconda를 이용한 설치는 별로 어렵지 않을 것이라고 생각했었다. 그런데 테스트 실행을 하는 과정에서 일찌감치 문제가 발생하였다. 개발자가 제공하는 Docker image를 사용함으로서 설치에 따르는 문제를 우회해 보려고 시도하였으나 이 역시 잘 되지 않았다. 지금 생각해 보니 Docker image와 reference database의 버전 문제가 아니었나 하는 생각이 든다.
첫 실행 단계에서 벌어지는 문제에 대하여 GTDB 포럼을 검색해 보니 numpy의 버전을 낮추라는 제안이 있었다. Conda 자체를 새로 깔아서 해결했다는 코멘트도 있어서 일단 가장 쉬운 방법을 택했다. 즉, 내 데스크탑 서버에 아예 다른 계정을 새로 만들어서 conda를 깨끗하게 설치하고 GTDB-Tk를 환경도 구축하였다. Mamba가 나온 뒤로 conda 생태계도 조금씩 세분화되는 것 같다. 물론 환경 구축이나 패키지 설치에는 mamba가 훨씬 편리한 것은 맞는데, 'mamba activate <env>'라는 명령어를 입력하는 것이 아직은 어색하다. 예전에는 mamba가 conda이 패키지 매니징 기능만을 대체한다고 생각하였는데 점점 그 영역을 넓혀 나가는 것으로 보인다. 이번에는 새 계정에 conda를 설치하기 위해 miniconda가 아닌 miniforge를 사용해 보았다.
그래도 여전히 GTDB-Tk의 테스트 실행, 즉 첫 단계인 'gtdbtk identify'(prodigal을 이용한 gene prediction 단계)에서 문제가 발생하였다. 화면으로 나오는 메시지를 잘 살펴보았다.
...
AttributeError: module 'numpy' has no attribute 'bool'.
`np.bool` was a deprecated alias for the builtin `bool`. To avoid this error in existing code, use `bool` by itself. Doing this will not modify any behavior and is safe. If you specifically wanted the numpy scalar type, use `np.bool_` here.
The aliases was originally deprecated in NumPy 1.20; for more details and guidance see the original release note at:
https://numpy.org/devdocs/release/1.20.0-notes.html#deprecations
...
numpy 버전을 1.20보다 낮은 것으로 바꾸어야 할 것만 같다. Bioconda를 이용하여 GTDB-Tk를 최신 버전으로 깔면 numpy는 1.24.4가 되는데 이 무슨 조화란 말인가? 다음 명령어를 이용하여 gtdbk-2.1.1 환경을 그대로 유지한 채 numpy를 다운그레이드하였다.
(gtdbtk-2.1.1) $ mamba install -c conda-forge numpy=1.19.5
numpy 버전을 내리고 나서 다시 gtdbtk 명령어를 실행해 보았다. 비로소 identify - align - classify로 이어지는 전 과정이 순조롭게 진행되었다. 화면으로 표시되는 진행 상황을 살펴보니 마커 유전자를 발굴하는 데에는 TIGRFAM protein family를 쓰는 것 같았다. 아, 추억의 TIGRFAM이여... 언제 기회가 된다면 미생물 유전체 분야의 값진 유산인 TIGRFAM에 대해서 글을 남겨야 되겠다.
최종 결과는 박테리아와 고세균 각각에 대해서 별도의 summary.tsv 파일(<prefix>.bac120.summary.tsv and <prefix>.ar53.summary.tsv)로 제공된다. 이 파일에 대한 설명은 여기를 참조하라. 결과 파일의 맨 마지막 컬럼인 'warning'(indicates unusal characteristics of the query genome that may impact the taxonomic assignment)에서 어떠한 글을 남겼는지 확인해 보았다. 72개 유전체 어셈블리 중 8개에 대해서 경고문이 나왔으며, 다음과 같은 세 부류로 압축된다.
- Genome has more than ##.#% of markers with multiple hits
- Genome not assigned to closest species as it falls outside its pre-defined ANI radius
- Insufficient number of amino acids in MSA (#.#%)
1번은 아마 다른 균주로 오염이 된 것일테고, 2번은 type stain과 비교 시 ANI threshold 이내에 들어오지 못하는 것으로 보이며, 3은 sequencing coverage가 충분하지 못하여 genome completeness가 떨어진 것으로 보인다. GTDB-Tk 분석을 하기 전 k-mer analysis, phyloFlash 및 ZGA pipeline을 통해서 read 및 assembly 차원에서 품질이 떨어지는 샘플를 추려 놓았으니 이를 GTDB-Tk의 결과와 비교해 보면 재미있을 것이다.
남들이 이미 만들어 놓은 멋진 도구나 자원을 늘 가져다 쓰기만 하는 것이 부끄럽다. 은퇴하기 전에(아직 꽤 많이 남았다!) 뭔가 나도 이 업계에 기여했다는 흔적은 남겨야 하는데...
챗GPT에게 anaconda, conda, miniconda, mamba 및 miniforge의 차이를 설명해 달라고 부탁해 보았다. anacond~miniconda~miniforge(배포판), conda~mamba(환경 및 패키지 관리 도구)라고 보면 쉽다. 다음은 챗GPT의 답변 일부를 인용한 것이다. Bioconda와 같은 '채널'을 이해하는 것도 중요하다.
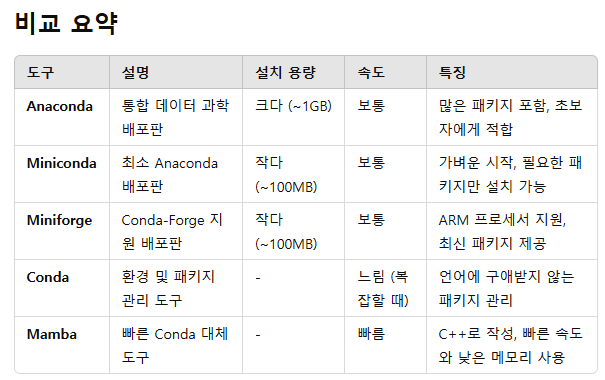
추천 사용 시나리오(챗GPT 답변)
초보자: Anaconda
데이터 과학 및 머신러닝을 빠르게 시작하고 싶다면 Anaconda가 적합합니다.고급 사용자: Miniconda 또는 Miniforge
최소 설치 후 필요한 패키지를 선택적으로 설치하려는 경우 Miniconda나 Miniforge를 권장합니다. 특히 M1 Mac과 같은 ARM 기반 환경에서는 Miniforge가 적합합니다.빠른 속도: Mamba
Conda의 느린 속도에 불만이 있다면 Mamba를 사용하는 것이 좋습니다. Conda와 동일한 명령어로 사용할 수 있습니다.
댓글 없음:
댓글 쓰기