유전체 서열 정보를 이용한 미생물 균주의 정확한 동정은 기초 생명과학과 감염병 관련 연구 분야에서 매우 중요한 일이다. 종(species) 또는 더 세분화된 수준으로 균주를 동정하거나 - 후자를 보통 타이핑(typing)이라고들 한다 - 유전체가 보유한 항생제 내성(AMR, antimicrobial resistance) 및 병원성 인자(VF, virulence factor)를 예측하는 것도 중요하다. 만약 유전체 해독을 실시한 미생물 균주가 쓸모있는 이차대사물을 만들어내는 것 같다면, 이를 책임지는 유전자 클러스터를 예측하는 것도 필요하다.
이러한 분석을 한꺼번에 실시하는 '입맛에 딱 맞는' 파이프라인은 아직 눈에 잘 뜨이지 않는다. 다만 병원성 미생물의 동정과 관련된 도구들은 가끔씩 눈에 보여서 이를 설치하고 테스트하면서 나름대로 평가를 하고 있다.
Frontiers in Microniology에 최근 공개된 fIDBAC라는 분석 플랫폼을 요즘 열심히 테스트하고 있다. fIDBAC는 면밀한 검토를 거쳐서 작성된 type strain 유전체 DB(N=12,784; 전체 목록은 12784.genome.txt를 참조)을 바탕으로 하여 미지의 유전체가 주어졌을 때 이것이 어떤 종에 속하는지를 정확히 판별해 주는 것을 목표로 한다.
- 논문 웹사이트(DOI=10.3389/fmicb.2021.723577)
- fIDBAC 웹사이트
- GitHub
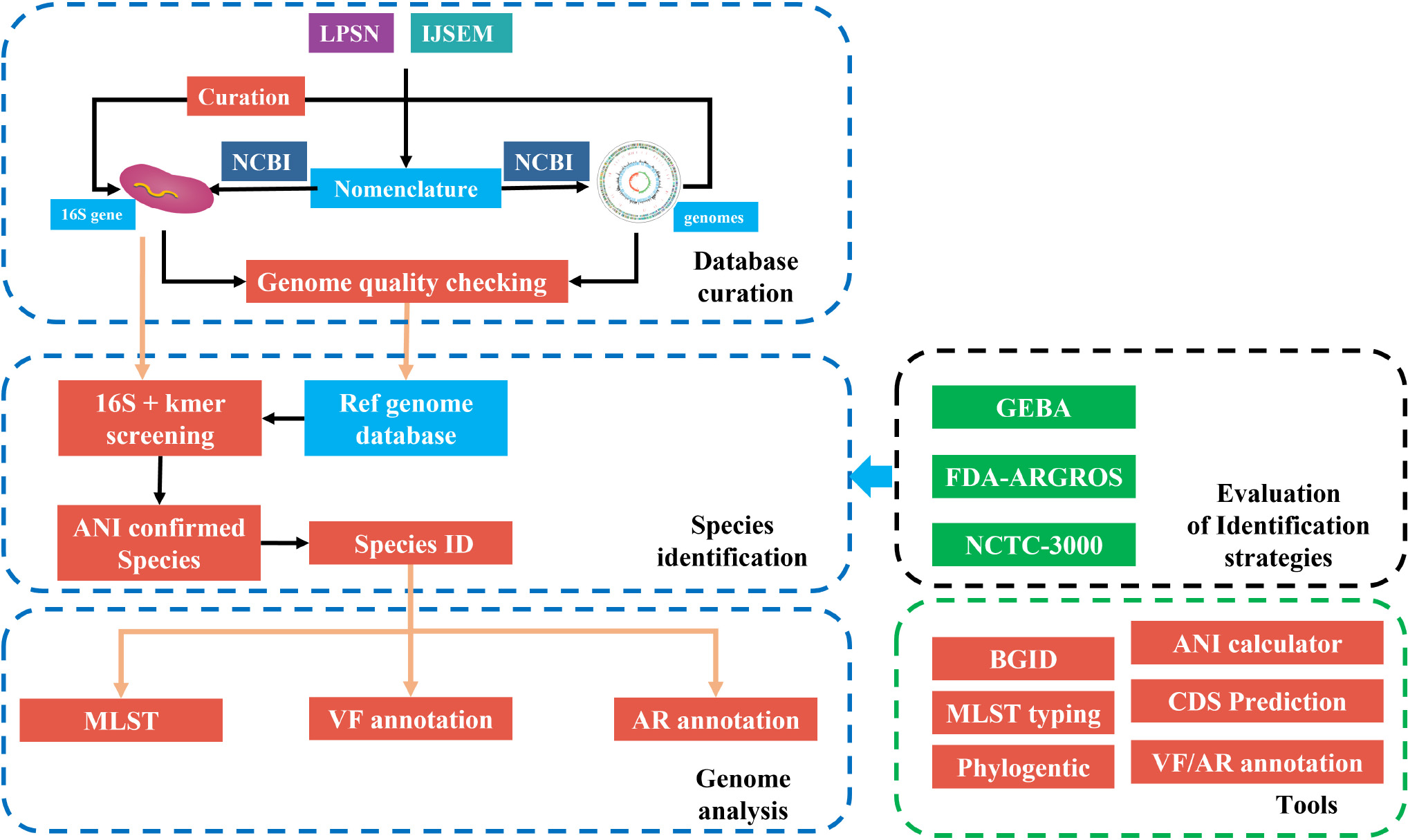 |
| fIDBAC의 프레임워크. 출처 링크 |
여기까지는 이름이 제대로 되어 있는지를 점검하는 것에 지나지 않는다. 다음 단계에서는 해당 균주의 유전체가 얼마나 온전한지를 확인하는 것이 필요하다. CheckM으로 점검하여 contamination이 5%를 넘거나 completeness가 90% 미만인 것은 버리고, 16S rRNA sequence를 추출하여 LTP database와 비교하여 genus level에서 불일치하는 것은 제외하였다.
SILVA, RDP, GreenGenes DB는 많이 들어 보았지만 LTP('The All-Species Living Tree' Project)는 생소하여 검색을 해 보았다. 이곳에서 LTP의 상세한 연혁 소개와 함께 다운로드 기능도 제공한다. 앞서 소개한 잘 알려진 ribosomal RNA database가 있음에도 불구하고 LTP가 별도의 프로젝트로서 존재해야만 했던 이유가 있었을 것이다.
큐레이션의 마지막 단계에서는 모든 유전체에 대한 pairwise ANI 계산을 실시하여 동일한 종명을 갖는 것끼리를 클러스터링하였다. 여기에서는 SciPy 파이썬 패키지를 사용하였다고 한다. 이렇게 만들어진 type strain의 유전체 염기서열은 KmerFinder를 통해서 k-mer (다운로드 링크)DB로 변환하였다. KmerFinder에 대한 상세한 정보는 관련 논문을 참조하라.
Reference DB 구축이 완료되었으니, 이를 활용하여 query genome을 동정하는 방법을 수립하는 것이 fIDBAC platform의 나머지 절반 과정이 되겠다. 메인 스크립트(fIDBAC.pl)을 통해서 작동 순서를 알아보면 다음과 같다. 이는 논문에서 소개한 순서와는 조금 다르다.
- Reference genome의 k-mer DB에 대하여 검색 실시(findTemplate without -w). 결과 파일은 $sample.out.txt이다.
- Genome annotation 실시(prokka) 후 16S rRNA와 유전자 서열 추출. 아미노산으로 번역한 유전자는 항생제 내성 및 병원성 인자를 탐색하는데 쓰임(AR_VF.run.pl)
- 16S rRNA에 대한 검색 및 fastANI를 이용하여 query genome의 종 동정 정보를 fastANISpecies라는 파일에 한 줄로 출력. 이것은 select_kmerfinder_16SAndANI.pl 스크립트를 통해서 이루어지는데 내부 구조가 좀 난해하다. [1]번 과정의 결과 파일($sample.out.txt)로부터 top1/3/10/20의 목록을 취한다. 최고 스코어를 보이는 genome이 3개라면 top1 목록은 세 줄이 된다. FASTA file 이름으로부터 목록 파일을 참조하여 taxonomic name을 얻는 과정이 꽤 된다. 16S rRNA 서열은 16S.V3.fa(관련 파일은 전부 여기에 있음; 이것이 LTP3132_SSU에서 유래한 것으로 추정됨)에 대하여 blastn을 실행하여 top 20을 얻은 뒤, local FASTA file에서 hit에 해당하는 것을 찾아 놓는다. fastANI에 계산에 투입되는 top 20은 16S 분석과 KmerFinder에서 얻어진 top genome을 추려서 얻어지는 것이 아닐까 생각하는데, Perl 스크립트를 뜯어보면서 정말 그러한지를 확인하지는 못하였다. 논문에서는 "The top 20 closest species were extracted from the results of the above-mentioned methods(즉 16S rRNA 서열 분석과 k-mer 분석)."이라고 하였으니 두 가지 결과를 종합하는 것이 맞을 것이다. 단, 두 가지 분석을 통해 얻어진 top 20 closest genome 안에서 종명이 consistent하지 않은 것이 섞여있을 경우 어떻게 처리하는지는 아직 잘 모르겠다.
- OrthoANI.all_tre.new.py 스크립트를 사용하여 ANI.pl 실행. 이 Perl wrapper script는 엄밀히 말해서 천랩이 개발한 OrthoANI와는 다른 것인데 왜 이런 이름으로 부르는지를 모르겠다. 이 wrapper script는 아직 잘 작동하지 않는 것 같다. 왜냐하면 주요 결과 파일인 OrthoANI.txtd와 ANI.all.txt가 빈 상태이기 때문이다. 왜 그런가 추적을 해 본 결과 이유는 너무나 허무한 곳에 있었다. OrthoANI.all_tre.new.py 스크립트 내부에 ANI.pl의 경로가 지정되어 있었다. 아니, config_db.txt 파일은 뭐하러 만들었단 말인가? 부글부글...
- ANIcaculator를 사용하여 gANI 계산. 유전자는 prokka가 만든 것을 이용한다. Step [4]와 [5]의 상호 연관성은 아직 파악하지 못하였다. ANI > 95%인 것만을 선별하는 것은 fastANI 레벨인가, gANI 레벨인가?
- MLST analysis
- SNP and MST analysis
- ....어휴!
치명적인 약점(?) 또는 개선할 점을 생각해 보았다. 우선 archaea는 레퍼런스 DB에 전혀 반영되어 있지 않다는 점을 들고 싶다. Archaea는 인체에 병을 일으키는 것이 없다고는 하지만, 일반적인 species 동정을 목적으로 fIDBAC을 이용하려는 사람에게는 치명적인 단점이다. 만약 고균을 포함한다면 fIDBAC은 fIDBAAC(...Bacterial and Archaeal...)이 되어야 할 것이다. k-mer DB는 보다 현대적인 Mash 기반 DB로 바꾸는 것이 바람직할 것이다. 12,000개 정도의 유전체 서열을 다운로드하여 k-mer DB(maketemplatedb 명령)을 어젯밤부터 실행했는데 아직 400개도 진행을 하지 못했다. 파이썬 2.7을 써야 하고(버전 3에서 작동하도록 고쳤다는데 잘 안됨), 다중 쓰레드도 쓰지 못하니 너무나 답답하다.
그래도 fIDBAC을 뜯어보면서 얻은 성과가 있다면 ReadSeq이나 phylip 등 고전적인 프로그램을 다룰 기회가 생겼다는 점이다.
댓글 없음:
댓글 쓰기